REFERENCE MANUAL
Note: click on the hyperlinks to navigate throughout the manual
For questions or suggestions, please contact F. Javier García de Abajo
| ELECTRON DIFFRACTION IN ATOMIC CLUSTERS (EDAC) | ||||
| for the simulation of Photoelectron Diffraction (PD) | ||||
|
REFERENCE MANUAL |
|||
Note: click on the hyperlinks to navigate throughout the manual |
||||
For questions or suggestions, please contact F. Javier García de Abajo |
||||
| INTRODUCTION |
EDAC is a multi-purpose code that implements multiple scattering (MS) of electrons in atomic clusters. This manual describes its capabilities
concerning core-level photoelectron diffraction (PD) in solids
and atomic clusters, including
relativistic effects (i.e., spin-flip during scattering and spin-orbit
coupling in the initial states), magnetic exchange effects
(the program is able to deal with two spin components separately),
and non-spherical atoms (this requires special input from the user
related to the atomic scattering matrices). Angular and energy scans can be performed with flexibility.
EDAC is written in C++ and it is rather machine independent.
It can be run in many platforms, including Unix, Linux, and
Windows, and it can be compiled with GNU g++ and SUN C++
compilers.
The cluster approach followed in this code consists in
representing the solid by a finite number of atoms N surrounding the emitter atom.
The finite inelastic mean free path limits the range of distances up to which the photoelectron
can travel within the solid without suffering inelastic collisions,
and this defines a finite volume which justifies the use of
an atomic cluster.
The muffin-tin model is assumed here, so that the atoms are
represented by their scattering matrices and they are considered
to be embedded in an homogeneous region of constant
potential. The atomic scattering matrices are completely
determined by the scattering phase shifts for spherical
atoms, but EDAC can deal with both spherical and non-spherical
atoms. In particular, the code can calculate scattering
phase-shifts internally for spherical atoms, provided
their so-called muffin-tin potentials are given in the input.
Also, it can calculate approximate muffin-tin potentials (and therefore atomic phase shifts and scattering matrices as well)
for specific cluster arrangements using information of free-atom
potentials (stored internally) and standard averaging procedures.
The MS expansion is performed up to an order n specified by
the user. Different iteration techniques are available in the code.
Overall, the computation time is proportional to
the number of complex multiplications that have to be performed,
and this in turn is Titer ~ nN2(lmax+1)3,
where lmax is the maximum value of the
orbital angular momentum number.
Alternatively, an exact solution of the self-consistent MS equations can be performed
in a time proportional to Texact ~ N3(lmax+1)6 (this is computationally very demanding and can be performed only
for small cluster sizes and small values of lmax).
The value of lmax is adjusted dynamically
during the calculation for each atom and each photoelectron
energy, so that negligible matrix elements or scattering
phase shifts are not used in order to avoid investing
computation time to calculate zeros.
For more details on the theory underlying these issues,
the user is referred to García de Abajo, Van Hove, and
Fadley, Phys. Rev. B 63, 75404 (2001).
Once the code has been compiled and an executable file named edac has been created, EDAC can be run simply by typing edac filename1 filename2 ... where filename1, filename2, ...
are input files containing commands to
be performed sequentially (normally, only one input file will be used).
EDAC input files are preferably given the extension .ms
(for multiple scattering),
so that both edac sample.ms and edac sample will use file sample.ms as input, although the latter option will use file sample is file sample.ms does not exist.
If no file is specified, the program starts in inline mode and the instructions are directly
read from the keyboard (this can be used within Unix systems in
combination with echo or as cat sample.ms | edac;
to exit this mode you simply have to type end).
The format of EDAC instructions is relatively flexible.
A detailed description is given below.
The input file has to contain information on the following
topics:
Atomic cluster. The cluster atomic coordinates and the type of atoms (i.e., their chemical nature or alternatively the type of scattering properties).
Mobility of beam and analyzer. How the sample moves with respect to the beam and the analyzer.
Energies and angles of emission. The range of energies and angles of emission of the photoelectrons.
Incident beam. The angle of incidence and polarization of the incoming light.
Emitters. The atoms that are emitters and their scattering properties when they are ionized (if various emitters are selected, the program calculates the sum of photoelectron intensities from each of them).
Initial core level. The initial core level from which the photoelectron is emitted. Also, whether the sum over sublevels is required.
Atomic scattering parameters. The scattering properties of the different atoms in the form of input files with scattering phase shifts, muffin-tin potentials, or scattering matrices. Alternatively, the muffin-tin potentials of the cluster atoms can be calculated internally (provided the atomic numbers or atomic symbols of those atoms have been specified) using the command muffin-tin.
Multiple scattering parameters. lmax, the type of iteration method, the orders of iteration, the muffin-tin zero, the temperature, and the electron inelastic mean free path.
Actual PD calculation. Invoke the command scan pd ... that performs the PD calculation.
The order of the different commands is to some extent arbitrary,
although many restrictions apply (e.g., a cluster must be defined
before invoking a PD calculation). Therefore, the structure
of the input file suggested by the above list is recommended.
The calculated far-field photoelectron emission intensities correspond
to the triply-differential energy-solid-angle distribution probability
per unit time in a.u., assuming that the external electric field has
an strength of 1 a.u.
The units of the rest of the input and output
magnitudes are explained below.
Finally, the MS calculation is performed for an atomic cluster
surrounded by an infinite space of constant potential. The value of the
potential outside the cluster is just the muffin-tin zero.
The effect of the surface is taken into account by introducing
a transmission factor (obtained from the transmission of an electron
through a step barrier of height equal to the muffin-tin zero)
and by transforming the directions of emission from/to the vacuum to/from
inside the solid via the corresponding refraction at the surface
barrier. A shift in energy corresponding the the potential step is also
incorporated.
Here are some examples of input files along with their
respective output (click on the chosen example for more details):
General sample of input file. This contains most of the common features of the program and all of the commands that are mandatory to run a PD calculation. It contains comments that indicate what can be changed to the convenience of the user.
A full list of commands in alphabetical order is included at the end of this document with links to more detailed
explanations and discussions.
| ATOMIC CLUSTER SPECIFICATION |
EDAC evaluates the MS series for a photoelectron in
the presence of an atomic cluster. The latter can be specified
either in an external output file or by means of commands that add/remove individual atoms, rows
of atoms, planes of atoms, or an entire semi-infinite crystal
to/from the cluster. These commands are explained below in detail.
The number of atoms that are stored in the program is controlled
by the parameter Rmax (see below). When an atom, row of atoms,
layer of atoms, or semi-infinite crystal
is added to the cluster, only the coordinates of those atoms that are
closer than Rmax to a given reference point
(xr, yr, zr) are actually stored in the cluster.
When running a photoelectron diffraction calculation, not all
the atoms stored in the cluster are actually used, but just those
whose distance to the emitter is less than the current
value of Rmax (the user may change this several
times during the execution of the program).
Furthermore, the maximum number of atoms used in
photoelectron diffraction calculations is also limited
by the command cluster natoms N.
Besides its coordinates, each atom is characterized by a label that
identifies the kind of scatterer that it represents. The meaning
and format of this label are discussed below.
| Cluster commands |
Adding and removing atoms, layers of atoms, and
semi-infinite crystals
Note: the parameters θmagnet and φmagnet are only required
when the flag orientation on has been invoked (it is off by default).
add cluster atom i units2 x y z [θmagnet φmagnet]
Add an atom of scatterer type i to the cluster at the position (x,y,z), where the coordinates are
given in units of units2.
If an atom already exists at that position, this
command just changes the atom type to i.
If the distance from (x, y, z) to the reference point
(xr, yr, zr) is larger than Rmax,
this command is ignored.
The parameters θmagnet and φmagnet specify the
orientation of the magnetic moment of the atom and they
have to be given if and only if the flag orientation on has been invoked. Only scatterers with non-negative i can be defined; negative i's refer to virtual atoms,
which do not scatter the electron: this can be used to
create sources of photoelectrons at places where there is
no physical atom.
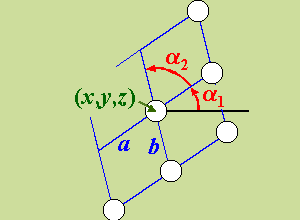
add cluster layer i units2 x y z a b α1 α2 [θmagnet φmagnet]
Add a layer of periodically ordered atoms of scatterer type i to the cluster. The layer is perpendicular
to the z-axis and the 2D unit cell is defined by two
vectors a and b of lengths a and b,
respectively (see figure).
The angle between the x-axis and
the first unit-cell vector (a) is α1.
The angle between the first vector a and the
second one b is α2.
The coordinates of the layer atoms are given by
(x +n acos(α1)
+m bcos(α1+ α2), y +n asin(α1)
+m bsin(α1+ α2), z)
in units of units2,
where n and m are integers.
All atoms of the (infinite) layer whose distance to the reference point
(xr, yr, zr) is less than Rmax will be added to the cluster.
If no value of Rmax has
been specified, the program stops.
The parameters θmagnet and φmagnet specify the
orientation of the magnetic moment of the atoms and they
have to be given if and only if the flag orientation on has been invoked.
 |
add cluster half-layer i units2 x y z a b α1 α2 φ [θmagnet φmagnet]
Add a half layer to the cluster.
The parameters of this command are the same as in add cluster layer ....
Only those atoms of the layer with coordinates
(xa, ya, za) that satisfy the condition
(x-xa) cos(φ)+
(y-ya) sin(φ)>=0
are added to the cluster.
Here, φ is the angle between the
half layer boundary and the x direction.
This command is useful to define clusters that represent
stepped surfaces.
add cluster row i units2 x y z a θrow φrow [θmagnet φmagnet]
Add a row of periodically arranged
atoms of scatterer type i.
The row is parallel to the polar
direction (θrow, φrow), with an atom at
(x, y, z)
and spacing a, both in units of units2.
All atoms of the (infinite) row whose distance to the reference point
(xr, yr, zr) is less than Rmax will be added to the cluster.
If no value of Rmax has
been specified, the program stops.
The parameters θmagnet and φmagnet specify the
orientation of the magnetic moment of the atoms and they
have to be given if and only if the flag orientation on has been invoked.
add cluster surface i units2 x y z a surface-type [c] [θmagnet φmagnet]
Add a semi-infinite crystal of atoms of scatterer type i.
One of the outer-most surface atoms is located at
(x, y, z)
and the lattice constant is a, both in units of units2, for sc, bcc, and fcc crystals,
in which case the parameter c does not have to be given.
For hcp crystals (see below) the parameter c has to be
given, and it refers to the distance between (111) planes, whereas a refers to the distance between nearest neighbors within
(111) planes rather than to the lattice constant.
The crystal extends towards the negative z region.
All atoms of the semi-infinite crystal whose distance to the reference point
(xr, yr, zr) is less than Rmax will be added to the cluster.
If no value of Rmax has
been specified, the program stops.
The surface is of type surface-type, which can take one of
the following values:
sc100, bcc100, fcc100: in this case x || [100], y || [010], and z || [001]
sc110, bcc110, fcc110: in this case x || [-110], y || [001], and z || [110]
sc111, bcc111, fcc111, hcp0001: in this case x || [11-2], y || [-110], and z || [111]
The crystal structure is of type simple cubic (sc), bcc, or fcc in each case (click on these words for graphical details). The special case of hcp(0001) surfaces is also included. The parameters θmagnet and φmagnet specify the orientation of the magnetic moment of the atoms and they have to be given if and only if the flag orientation on has been invoked. Diamond, NaCl, and other types of lattices can be built as a superposition of sc, bcc, and fcc lattices.
remove cluster atom units2 x y z
Remove an atom from the cluster at the position
(x, y, z)
(this has no effect if there is no atom at that position).
If |(x, y, z)-
(xr, yr, zr)|
is larger than the current value of Rmax, this command
is ignored.
remove cluster layer units2 x y z a b α1 α2
Remove atoms from a cluster layer. The sites of
the layer atoms selected by this command are the same as in the command add cluster layer ... with the same parameters.
If no value of Rmax has
been specified, the program stops.
remove cluster row units2 x y z a α1 α2
Remove the atoms that are at the position of those included by cluster add row ... with the same parameters.
If no value of Rmax has
been specified, the program stops.
clear cluster
Remove cluster coordinates and list of emitters.
Limiting the number of atoms
cluster natoms N
Set the maximum number of atoms N that can be included
in actual calculations. If this is smaller than the
number of currently stored cluster atoms,
the program selects N atoms chosen as close as possible
to the emitter(s) or the reference point when running the commands scan pd ... or cluster output ...,
respectively. The flag cluster surface on/off can be relevant here.
Use N=all to include all stored atoms. Caution: Limiting the number of atoms using this command
can break the symmetry of the cluster, and therefore, also
the symmetry of the diffraction pattern. Default: cluster natoms all.
cluster reference-point units2 xr yr zr
Set the values of the coordinates of the reference point
(xr, yr, zr).
This is not required to coincide with any cluster atom. Default:
(xr, yr, zr)=
(0,0,0).
cluster Rmax units2 Rmax
Set the maximum distance Rmax from the emitter (while running a PD calculation)
or from the reference point (while writing the cluster to a file or while running add cluster ... or remove cluster ... commands)
up to which atoms are considered to belong to the cluster.
Atoms further away are ignored.
The command cluster surface on/off determines the actual shape of the selected cluster (see figure
below, where the red atom acts as the reference point). Default: Rmax=infinity.
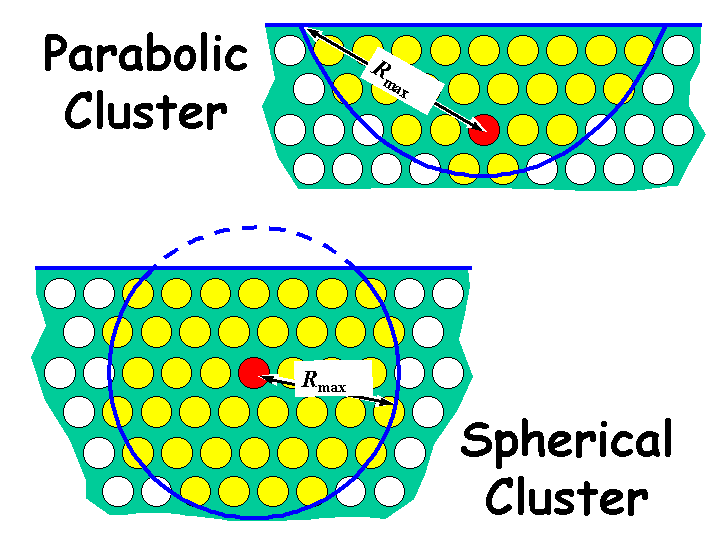 |
cluster surface on/off
Either the keyword on or off must be used.
When this flag is off, the definition of distance between
atoms i and j used to compare with Rmax has the usual geometrical meaning
|ri-rj|.
However, when this flag is on that distance is redefined as
|ri-r0|
+ (zsurf-zi),
where zsurf is the position of the surface
defined by the command cluster surface zsurf and r0 is either the reference point or an emitter.
When the number of atoms is restricted by either cluster Rmax units2 Rmax or cluster natoms N and
this flag is on, the shape of the atomic cluster actually
selected has a parabolic shape with its focus at the position
of the emitter atom (in PD calculations)
or at the reference point (rest). Default: off.
cluster surface units2 zsurf
The surface is taken at z=zsurf in units
of units2.
This controls the inelastic attenuation
of the photoelectrons before escaping from the sample. See also cluster surface on/off. Default: zsurf=0.
Displacing and rotating the cluster
displace cluster units2 x y z
Displace all the atomic coordinates of the cluster by adding to them
the vector (x, y, z).
rotate cluster x/y/z φ
Perform a rotation around the axis x, y, or z (the axis of rotation passes by the origin (0,0,0) in all cases).
The angle of rotation is φ.
For φ>0, the rotation is
counter-clockwise as observed from the positive direction
of the corresponding axis. Example of use: rotate cluster z 30. This rotates
the cluster 30 degrees around the z axis
in the counter-clockwise direction.
Reading/writing cluster from/to a file
cluster input filename
Discard any previous information on the
cluster coordinates and read new cluster
coordinates from file filename with a specific format.
cluster output units2 filename
Print out the coordinates of the first N atoms
selected by the command cluster natoms N (i.e., those that are closer than Rmax to the reference point
(xr, yr, zr))
in file filename (if no value of N has been specified, it prints out all
stored atoms).
They are written in units of units2.
The format of this file is the same as in cluster input file,
so that it can be used as input with that command.
Miscellaneous
report natoms
Print current value of N selected by the command cluster natoms N fixed either by the user
(if no calculation has been performed after using
the commands cluster natoms N or cluster Rmax units2 Rmax)
or by the last PD calculation (otherwise).
This shows the total number of atoms stored in the cluster as well.
| Cluster data-file format |
The file read by cluster input filename or written by cluster output filename has the following structure:
N units2 |
| L1 T1 x1 y1 z1 [θ1 φ1] |
| L2 T2 x2 y2 z2 [θ2 φ2] |
... |
| LN TN xN yN zN [θN φN] |
where N is the number of atoms in the cluster,
the coordinates
(xj, yj, zj)
are given in units of units2, Lj are atomic
labels (1 through N without repetition),
and Tj is the type of scatterer i that atom j represents.
The parameters θj and φj specify the
orientation of atomic magnetic moments (if the atom
in question has a magnetic moment at all) and they
have to be given if and only if the flag orientation on has been invoked (it is off by default).
The actual scattering properties of each atom are specified by the command scatterer Tj ... and properties for all Tj's must be defined before a PD
calculation is performed.
Alternatively, Tj can be either an atomic number
or an atomic symbol, and this must be done in combination with
the command muffin-tin (click here for further details).
The labels Lj change when performing calculations
or when writing the cluster to a file. However, they can
be used immediately after reading a cluster file to
specify the list of emitters via the command emitters n i1 L1 ...
Example of cluster file (for non-oriented atoms): oxygen (blue atoms)
on top of tungsten (red atoms):
|
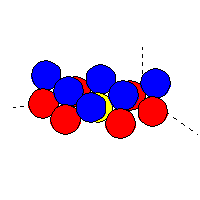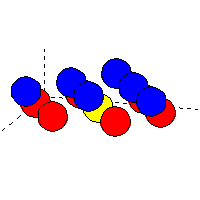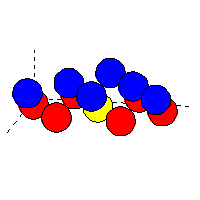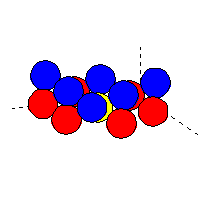 |
The yellow atom (tungsten) is at the position (0,0,0).
The scatterer type labels (74 and 8 in the example) are changed when the command muffin-tin is
invoked to construct the muffin-tin potentials.
The command cluster output ... can be used after that to see what the new labels are.
| DESCRIPTION OF THE BEAM AND THE ANALYZER |
The angles of incidence of the incoming photon beam and the angles
of emission of the photoelectron are represented in the following figure:
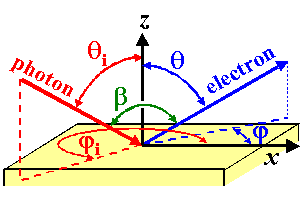 |
Besides the mode of mobility, the input
file has to specify the electron energy range
and angular range of emission and the beam incidence direction and polarization.
Next, a description of the modes of angular scans that are supported by
the code is given, along with an explanation of the relevant commands.
| Mobility of beam and analyzer |
Various different general modes of motion of detector and sample are supported by this code. In all of them, the polar emission angle θ refers to the polar direction of the detector with respect to the surface normal (the z-axis of the cluster), and the azimuthal emission angle φ refers to the azimuthal direction of the detector with respect to the x axis (all atomic positions are expressed in terms of their Cartesian coordinates (x, y, z)). A detailed description of each mode of motion follows:
Move only the sample: In this mode, the angle between
the incoming beam and the analyzer is kept constant and equal
to the input parameter β.
The incidence angles θi and φi (defined by incidence ... commands) are ignored. The sample moves in such a way
that the surface normal is always contained in the plane determined
by the incidence and emission directions, and the polar angles of
emission are θ=β and φ=0 for normal incidence.
The azimuthal angle is measured with respect to the x axis.
This mode is selected with the command movable cluster (see figure below).
Move only the analyzer (the cluster is fixed): In this mode, the incoming beam is fixed
with respect to the sample, so that by varying the angles of emission θ and φ, only
the analyzer moves.
The incidence direction is kept constant with respect to the sample,
so that the parameter β is ignored.
This mode is selected with the command fixed cluster (see figure below).
Move both the sample and the analyzer: In this mode, the sample moves with the azimuthal angle of emission φ, while the analyzer moves with
the polar angle of emission θ.
The direction of incidence
(i.e., from where the photon beam is coming)
with respect to the surface normal
is (θi, φ+π)
when the direction of emission
is (θ,φ).
The azimuthal angle of incidence φi and the angle β are ignored.
This mode is selected with the command semi-movable cluster (see figure below).
Moreover, some specific geometries for particular beamlines have been implemented (beamline geometries that are not contained in this list can be implemented upon request):
Beamline 7.0.1 of the ALS synchrotron (Berkeley).
The polar angle of emission θ is referred to the surface normal (i.e., the z axis) and the
azimuthal angle of emission φ is relative to the x axis. The polar axis of rotation
is the y axis. Positive azimuthal rotations
(i.e., positive φ's)
are clockwise as observed from the surface normal.
Positive polar rotations are also clockwise when observed from
the direction opposite to the beam.
(See ALS web site for further details.)
This mode is selected by using the command beamline ALS-7.0.1.
Aebi's set up. This is similar to the movable cluster mode, but the beam, the analyzer,
and the surface normal will not be in the same plane.
See the following sketch for more details:
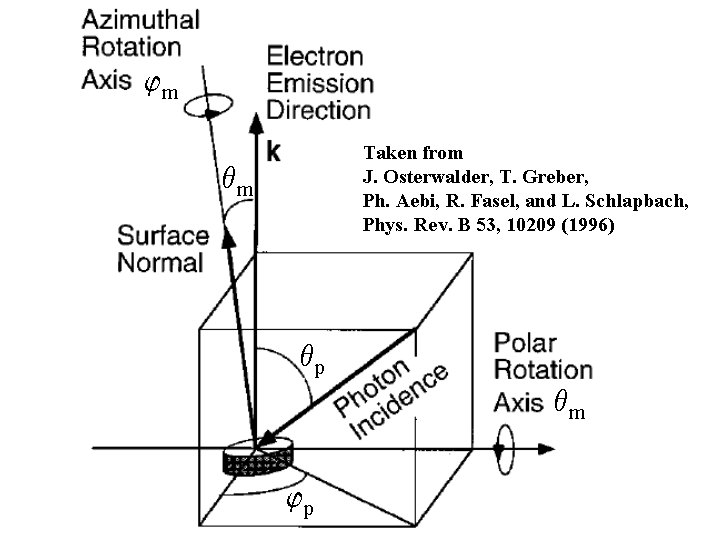 |
The emission angles selected
in the input file will correspond to rotations of the
manipulator as indicated in the figure. These angles are
therefore the polar direction with respect to the surface
normal. However, angles of incidence must be given
with this command that specify the polar direction of the photon
beam with respect to the electron analyzer,
(θp, φp).
This mode is selected by using the command beamline Aebi θp φp (p stands for photon).
The following schematic representation can help to visualize this.
Azimuthal/polar rotations are represented by brown/pink double arrows.
The sample and the analyzer move when these arrows are sitting on the
analyzer (blue arrow) and near the surface normal (black line),
respectively.
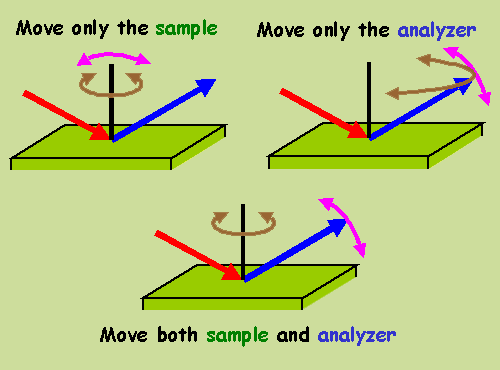 |
The commands that control the mobility of the beam and the
analyzer during angular scans are:
beta β
Set the angle between the direction
of the incoming light and the direction
of scattering/emission β (see figure above).
This parameter is only effective when the movable cluster option has been selected, and it is ignored otherwise. Default: β=0.
fixed/movable cluster
The cluster does not/does move when changing the
direction of emission
(θ, φ).
See the command beta β and
the schematic figure above.
With movable the angle between incident beam
and analyzer is kept constant
and equal to β;
everything happens as though only the target
moved when changing the direction of emission;
the polar angles of emission for normal incidence are
(β,0)
with respect to the surface normal (i.e., the positive z-axis).
With fixed, only the analyzer moves and the target is fixed
with respect to the incident beam. Default: fixed cluster.
semi-movable cluster
The angle between incoming beam and analyzer
is not kept constant, and θ refers to
the analyzer angle with respect to the surface
normal (positive z-axis). (See figure above.) The cluster rotates around z with φ, and φ=0
stands for the analyzer lying in the x-z plane
toward x>0. Default: fixed cluster.
beamline ALS-7.0.1
Uses the geometry of beamline 7.0.1 of the ALS (Berkeley). See above for further details.
beamline Aebi θi φi
Uses the geometry of Aebi's set up. See above for further details.
The last mode from this list that has been given at a certain point in the input file is the one that is active after that point.
| Energy and angle scanning parameters |
The energy range of the photoelectron and the angles of emission are specified by means of the following commands:
emission angle normal
Set the polar and azimuthal angles of emission to θ=φ=0.
emission angle phi φi φf nφ
Specify the azimuthal angle of emission φ, which takes values between φi and φf, and nφ is
the total number of equally-spaced φ angles. Default: nφ=0.
emission angle theta θi θf nθ
Specify the polar angle of emission θ, which takes values between θi and θf, and nθ is
the total number of equally-spaced θ angles. Default: nθ=0.
emission angle window Δ
This can be used to simulate a finite acceptance angle in
the photoelectron detector. The half-width angle of the
acceptance cone is given by Δ. Default: Δ=0.
emission energy units1 ki kf nk
Specify the range of photoelectron energy scanned by the analyzer, where ki and kf are the initial and final
energy (or momentum) in units of units1, nk is the total number of equally-spaced
energies (if units1 is energy) or
momenta (if units1 is momentum).
The energy (momentum) is given relative to vacuum, and this
differs from the value inside the solid if a muffin-tin zero potential has been specified. Default nk=0.
| Incident beam characterization |
By default, the incidence is normal to the surface and the polarization
is linear along the x axis. This can be changed to any direction
of incidence and light polarization using the commands described below.
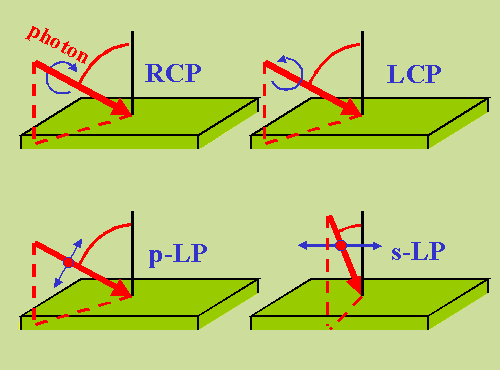
The polarization can be any of those represented in the following figure:
 |
Here are the commands that can be used to specify the direction of
incidence of the light and its polarization:
incidence θi φi
Set the values of the polar direction
(θi, φi)
of the beam (i.e., from where the photons are coming). Default: θi= φi=0. (See figure above.)
polarization α δ
Choose arbitrary polarization of the incident light
described by angles α and δ.
The light polarization vector is given by
[eθ cosα +eφ sinα exp(iδ)],
where eθ and eφ are unit vectors normal to the direction of
propagation of the light and parallel to the
polar and azimuthal polar directions, respectively. Default: α=δ=0.
polarization direction θε φε
Choose incoming linearly-polarized light defined by a
real polarization vector parallel
to the polar direction
(θε,φε).
polarization LCP/RCP
Chose left/right circularly-polarized light
(α=π/4, δ=π/2 for LCP
and δ=-π/2 for RCP). (See figure above.)
polarization LPx/LPy
Choose linear polarization with the polarization
vector parallel/perpendicular to the plane of scattering, which is
determined by the direction of the light and the surface normal
(i.e., δ=0, α=0 for LPx and α=π/2 for LPy). LPx and LPy correspond to p and s polarization,
respectively (see figure above).
| Spin-polarized detector |
The calculations performed by the program are done for unpolarized electrons when the scatterers and emitters do not include any electron-spin information. However, when any of the scatterers incorporates magnetic exchange effects or spin-orbit coupling, or when any of them is defined by an externally introduced t-matrix with two electron-spin components, or when the initial core sub-levels include information on electron spin (that is, by selecting a given spin using initial ms0 ... or initial mj0 ...), then the PD calculations performed by the program are done for the two spin components separately. The resulting photoelectron intensity is the sum of the two spin intensities, unless a spin-polarized detector is simulated using the following commands:
spin θspin φspin
Specify the polar angles of the direction on which the
detector projects the photoelectron spin.
This requires a previous call to spin on. Default: θspin= φspin=0.
spin on/off
Use spin on to make the detector project the electron
spin on the direction specified by the command spin ....
This command has no effect for non-polarized
PD calculations. Default: spin off.
| EMITTER ATOMS AND INITIAL CORE LEVELS |
The atoms that act as emitters must be specified
(they must all have the same chemical identity).
Then, the photoelectron intensities calculated in this code
represent the incoherent sum of the emission from all of
the selected emitters, with those closer to the surface
contributing more because of the smaller attenuation originating in
inelastic scattering.
An arbitrary number of emitters can be specified
(see incompatibilities with the keyword amplitude in the command scan pd ...). Each emitter is
characterized both by its coordinates and by
a label that specifies the type of scatterer
that represents it when it is ionized (different in general from
the non-ionized atom). When muffin-tin is invoked to calculate
the scattering properties of the cluster atoms internally (that is,
without the help of external data files) or when the atomic scattering
properties are given in terms of a muffin-tin potential, the latter label
is disregarded and a screened Coulomb hole is added to
the muffin-tin potential in order to calculate the scattering
properties of the ionized atom. The length of screening of the hole
is 1 Å by default, but it can be changed using the command screening-length ...
Next, the initial core levels and sublevels have to be specified.
This can be done in two different ways: either by selecting
the initial state (e.g., 1s, 2p, etc.),
so that the emission matrix elements are calculated internally
within the program, or by reading radial matrix elements from an external data file. In both cases,
a finer selection of sublevels can be achieved
by specifying further initial-state quantum numbers.
| List of emitters |
The following commands can be used to specify which atoms are emitters:
emitters all t1 t2
Make all atoms of type t1 to be emitters,
with scattering properties of type t2 when they are ionized.
The parameter t2 is ignored
when t1 refers to a scatterer that
is defined by its muffin-tin potential
(that is, when the command muffin-tin has been used or when a muffin-tin potential data file has been read to define it),
in which case the scattering properties of the
ionized atom are obtained from the muffin-tin
potential plus a screened Coulomb hole, as
explained above.
Moreover, t1 and t2 must be any of the labels introduced by scatterer t1 ...,
or alternatively, any of the scatterer-type labels created
when invoking the command muffin-tin (in this latter case, these labels can be obtained by writing the cluster to a file after muffin-tin has been invoked). Default: no emitters.
emitters n i1 t1 ... in tn
Add n emitter atoms to the list of emitters, where i1 ... in are the atomic labels of these new emitters and t1 ... tn are the corresponding type of
scatterers when they are ionized.
This command can only be invoked when (and immediately after)
the cluster is read from a file using the command cluster input ... The atomic labels i1 ... in are the same as those of the first column of
the cluster data file.
The scatterer-type labels t1 ... tn must be any of the ones introduced by scatterer t1 ... Default: no emitters.
emitters n units2 x1 y1 z1 t1 ... xn yn zn tn
Add n emitter atoms to the list of emitters. These
atoms are specified by their coordinates
(x1,y1,z1) ...
(xn,yn,zn)
in units of units2.
The program will stop if there are no atoms defined in the cluster
at all of these positions
(the tolerance in the input coordinates of the emitters is 0.05 Å).
The labels t1 ... tn are the corresponding type of
scatterers when the atoms are ionized, and they
must be any of the ones introduced by scatterer t1 ...,
or alternatively, any of the scatterer-type labels created
when invoking the command muffin-tin (in this latter case, these labels can be obtained by writing the cluster to a file after muffin-tin has been invoked). Default: no emitters.
report emitters
Print out current list of selected emitters.
This can be particularly useful to find the scatterer-type labels assigned by the command muffin-tin,
so that these labels can be used later to
extract phase shifts, muffin-tin potentials, etc.
Sometimes it is convenient to run the code a first time
in order to obtain those labels using report emitters and then end to stop the execution of the program,
and a second time (after utilizing those labels)
commenting out the end command.
| Initial core levels: n0 and l0 |
The following commands permit selecting the initial core-level
quantum numbers and calculating or inputting from external data
files the corresponding emission radial matrix elements.
Emission matrix elements calculated internally for a specific level
The emission matrix elements are calculated internally by default,
provided the emitter under consideration has been
defined by its muffin-tin potential (e.g., by invoking
the command muffin-tin).
The following commands can be used to specify the desired emission
level.
initial n0/l0 ν
Initial core-level principal and orbital quantum numbers.
Either the keyword n0 or l0 must be used
and the actual value is specified by ν
When using initial l0 1, for instance, both
2p1/2 and 2p3/2 components
(if the orbital momentum representation has been selected
using the command initial ljmj;
here, ljmj is a keyword and not a set of numbers)
or 2pspin-up and 2pspin-down components
(if the spin representation has been selected
using the command initial lmms)
are included.
The sublevel can be further specified by
using initial j0/mj0 ν or initial m0/ms0 ν,
respectively. Default: n0=1 and l0=0.
initial state label
This is an alternative to initial n0/l0 ν Labels such as label= 1s, 2s, 2p, etc.,
switch on the flag initial lmms and assign the corresponding value to n0 and l0.
Labels such as label= 2p1/2, 2p3/2, 3d3/2, etc.,
switch on the flag initial ljmj and assign the corresponding value to n0, l0, and j0 (previous calls to initial j0 ν are ignored). Default: 1s.
initial state C-R nt n1 X1 C1 n2 X2 C2 ...
Use the tabulation of Clementi and Roetti
(E. Clementi and C. Roetti, Atomic Data and Nucl. Data Tables 14, 177-478 (1974))
for the initial state.
When using this command, the radial matrix elements are calculated
using the coefficients given in the parameter list, where nt is the number of terms, n are the main quantum
numbers of the Slater functions, X are the coefficients
inside the exponentials (they dictate how far the wave function
extends), and C are the coefficients of each term in the
wave function. Notice that the initial quantum number l0 (and possibly m0) has
to be introduced separately.
Emission matrix elements defined via radial matrix elements
As an alternative to initial state ... or initial n0/l0 ...,
the emission matrix elements can be introduced via an external
data file whose format is specified below.
Here are the relevant commands in that case:
rmat filename
Read radial matrix elements from file filename.
The principal quantum number n0 becomes
unnecessary in this case, and the number l0 is specified inside the data file.
By default, the radial matrix elements are calculated internally
inside the code, except when this command is invoked, after
which they are simply read from the file filename.
clear rmat
Remove stored information on radial matrix elements.
Radial matrix elements are thereafter calculated internally
inside the code.
| Initial core sub-levels: spin and mj0 |
The following commands permit selecting the initial core sub-level
quantum numbers and the kind of representation that is used.
Two different representations can be used to specify
further the sublevel of emission. The first one
(default)
is the (lmms) or spin representation,
in which ms is the electron spin.
This can be selected by using the following commands:
initial lmms
Use the spin (lmms)
representation in the definition of the emission matrix
elements. lmms is a keyword and not a set of numbers.
Also, this de-activates initial ljmj. This is the default representation.
initial m0/ms0 ν
Initial core sublevel of emission.
This requires a previous call to initial lmms.
One has m0=ν for m0
and ms0=ν/2
for ms0 (i.e., ν=1 for spin up and ν=-1 for spin down).
Each sublevel involves a different initial state, and therefore
a separate MS calculation.
The resulting emission intensities
are simply added up. The input parameter ν can take the value all,
indicating that the full range
is selected (e.g., initial m0 all makes the selection m0=-l0, ..., l0). Default: m0=all and ms0=1/2.
A second representation available within this code is the (ljmj) or orbital momentum representation, which can be selected using the following commands:
initial ljmj
Use the angular momentum (ljmj)
representation in the definition of the emission matrix elements. ljmj is a keyword and not a set of numbers.
Also, this de-activates initial lmms. Default: initial lmms.
initial j0/mj0 ν
Initial core level and sublevel of emission.
This requires a previous call to initial ljmj.
One has j0=ν/2 for j0 and mj0=ν/2 for mj0.
Each sublevel involves a different initial state, and therefore
a separate MS calculation.
The resulting emission intensities
are simply added up. The input parameter ν can take the value all,
indicating that the full range
is selected (e.g., initial mj0 all makes the selection mj0=-j0, ..., j0). Default: all.
| Radial-matrix-elements data-file format |
The general form of the file that contains the radial matrix elements of a given emitter is:
| nk units1 index l0 |
| k1 a11 a12 a13 ... |
| k2 a21 a22 a23 ... |
| ... |
| knk ank1 ank2 ank3 ... |
where nk is the number of energies (or momenta), for which the radial matrix elements are given, units1 are the units in which the energies (or momenta) are given, k1, k2, ... are the respective energies (or momenta), either in decreasing or decreasing order (linear of cubic spline interpolation is used latter on for values lying in between), and a21, a22, ... are the data necessary to specify the radial matrix elements, which depend on the parameter index. The latter can be one of the following:
regular1. Each line for each energy (or momentum) kj contains:
|Rl0+1| δ l0+1 |Rl0-1| δ l0-1
(i.e., this is the information denoted aj1, aj2, ... above).
The radial matrix elements are Rl0+1 exp(iδl0+1)
for the l=l0+1 channel and Rl0-1 exp(iδl0-1)
for the l=l0-1 channel.
The commands initial m0 ... and initial ms0 ... can be used to select the initial quantum number m and the spin of the photoelectron (before MS) in the
(lmms) representation
(notice that these two commands require a previous call to initial lmms).
Also, the commands initial j0 ... and initial mj0 ... can be used to select an initial state that takes into
account spin-orbit coupling in the
(ljmj) representation
(notice that these two commands require a previous call to initial ljmj).
regular2. This is like regular1, but with the format:
Re{Rl0+1} Im{Rl0+1} Re{Rl0-1} Im{Rl0-1}
ex1. This can be used to incorporate magnetic exchange effects in the excitation matrix elements. Now each line for each energy (or momentum) contains:
|Rul0+1| δu l0+1 |Rul0-1| δu l0-1 |Rdl0+1| δd l0+1 |Rdl0-1| δd l0-1
where u and d refer to spin up and spin down
components, respectively.
In this case, previous calls to commands initial ... are ignored,
except initial m0 ... (initial ms0 ... is also ignored).
The option initial lmms is automatically selected when reading the file.
ex2. The same as ex1, but with the format of regular2.
An important part of the input data file is
the orbital angular momentum number l0 of the initial core level. Previous calls to commands initial n0 ... and initial l0 ... are ignored.
Here is an example (for l0=1, that is, emission
from a p state):
| 6 k(au) regular1 1 4 0.38 0.31 0.18 -0.01 5 0.30 0.40 0.16 -0.08 6 0.21 0.65 0.12 0.09 7 0.16 0.73 0.10 0.15 8 0.12 0.69 0.11 0.26 9 0.10 0.52 0.09 0.32 |
This is another example: 1 regular1 0 0.1 0.3 0 0.
The latter illustrates the special case nk=1, for which units1 and k1 do not have to be given.
Moreover, the l=l0-1 channel does not contribute
when l0=0.
Notice that 2π jumps in the phase of the
radial matrix elements are automatically eliminated while
they are read in order to avoid possible interpolation anomalies.
| ATOMIC SCATTERING |
Each of the atoms included in the atomic cluster must have a label that indicates the type of scatterer under consideration.
These labels can be given either as the atomic numbers
of the atoms or as their atomic symbols.
Then, the command muffin-tin can be invoked
after defining the cluster.
This command asks the program to calculate the muffin-tin potential
associated to each different kind of atom (identified by its atomic
symbol or atomic number, as specified in the definition of the cluster)
within the cluster (with the limitation imposed by Rmax).
The resulting muffin-tin potentials are stored
at that particular moment, so that they
are available to be used to obtain scattering phase shifts when these
are needed during the execution of an
actual PD calculation, performed later on using the command scan pd ... The muffin-tin potentials are automatically
calculated by the code using a muffin tin construction based upon
the Mattheiss prescription (Mattheiss Phys. Rev. 133, A1399 (1964)).
Atoms with different atomic number or
with the same atomic number but with different atomic environment (e.g.,
surface atoms versus bulk atoms) will have different muffin-tin potentials.
The cluster should include atoms that surround the
actual cluster used in the MS calculations that might follow, so that cluster-boundary
effects do not affect the actual cluster.
Notice that the scatterer properties of an emitter atom
are different when it is ionized as compared to the non-ionized atom,
since the former contains the photoelectron hole. This is incorporated
in the calculation by adding to the muffin-tin potential of the emitter
atom a point-like Coulomb hole which is screened with an exponential screening-length prescribed by the user
(1 Å by default).
While the procedure just described is convenient and reduces drastically
the input that the user must provide, one can still define the
scattering properties using external files (this is recommended only
for expert users that want to deal with special cases).
This is done by giving integral numbers (unconnected to the
atomic numbers) as input for the atomic labels
that refer to the type of scatterer under consideration.
Then the scattering properties for each kind of scatterer must be
read from external data files using commands that will be
explained below. In this case,
the effect of the photoelectron hole can be included
by defining scatterer properties for the ionized atom (different
from those describing the non-ionized atom) during the
selection of the emitters.
The screening length defined by the user does not apply here,
since the scattering properties of the ionized atoms are obtained
from an external data file.
| Basic commands |
At any given point in the input, the general properties of a given scatterer can be obtained using the following command:
Calculating the scattering properties inside the code
muffin-tin
Calculate muffin-tin potentials and muffin-tin radii
for the atoms of the cluster.
This command removes all information previously stored by means of scatterer ... The muffin-tin potentials are calculated only for the N atoms selected by the command cluster natoms N that are closest to the reference point.
The cluster should include atoms that surround the
actual cluster used in the MS calculations that might follow, so that cluster-boundary
effects do not affect the actual cluster.
The calculated muffin-tin potentials are
referred to the vacuum level. Later on, the
phase shifts corresponding to these potentials are calculated
whenever they are needed.
When using this command, atomic labels in the input of the
cluster definition will be interpreted as atomic numbers (e.g.,
29 or Cu).
After muffin-tin has been invoked, those labels
are changed to refer to the different muffin-tin potentials
that have been created. The new labels can still be
obtained by writing the cluster to a file using cluster output ...
screening-length units2 λscr
Set the value of the screening length
(λscr in units of units2) used to screen the photoelectron
core hole.
This command has an effect only when the scatterer
properties of the emitter are defined via its muffin-tin potential.
A screened-hole potential given by
-exp(-r/λscr)/r
is added to the emitter atom (the minus sign means that the hole
attracts the photoelectron). This represents in a
approximate way the presence of the localized core hole,
screened by the rest of the solid with the specified
screening length. Default: λscr=1 Å.
scatterer i sphere units2 a units1 V
Set scatterer i to represent a homogeneous sphere of radius a and constant potential V below the muffin-tin zero (the potential inside the sphere is -V with
respect to the muffin-tin zero).
Reading/writing scattering properties from/to external data files
scan scatterer i filename
Calculate the total (angle-integrated) elastic
scattering cross section (and the Sherman function
for relativistic phase shifts)
corresponding to scatterers of type i for the energies and angles previously specified by emission energy ... and emission angle ...,
respectively. This gives the angular dependent elastic
cross section, not
the total cross section.
The last three columns of the output correspond to
|f|2 (squared
modulus of the scattering amplitude f),
Re{f}, and Im{f}, respectively
(for relativistic phase shifts, the last three columns
correspond to |f|2, the Sherman function S, and |f|2S,
respectively).
The results are written in filename.
This command has no effect on scatterers defined
by an external t-matrix.
scan scatterer i phase-shifts filename
Calculate scattering phase shifts up to l=lmax for scatterers of type i within the energy range specified by emission energy ....
The results are written in filename,
which has a format compatible to be used as input of scatterer i filename (no results are written to the standard output).
This command has no effect if the scatterer is defined
via its scattering t-matrix,
but it is specially designed for
scatterers defined via their muffin-tin potentials, either by reading
them from a data file or by invoking the
command muffin-tin.
These are "temperature-dependent" phase shifts that
include the effect of the Debye-Waller factor: they
are calculated at the temperature
specified by temperature ... The potential stored for this scatterer is assumed to be referred
to the muffin-tin zero.
scan scatterer i potential filename
Write scattering muffin-tin potential of
scatterers of type i to file filename,
which has a format compatible to be used as input of scatterer i filename.
This command has no effect if the scatterer is not defined by
its muffin-tin potential, either by reading
it from a data file or by invoking the
command muffin-tin (no results are written to the standard output).
scatterer i filename
Read the phase shifts, the muffin-tin potential,
or the scattering matrix corresponding to scatterers of type i from file filename.
The kind of scatterer i is in correspondence with the labels
used to define the cluster atoms
(see add cluster atom ...).
The format of the input file is described below.
clear scatterer i
Remove scattering data from scatterer type i (phase shifts, etc.).
Notice that scatterer i ... must have been invoked before.
clear scatterers
Remove scattering data from all scatterers.
Spin-orbit coupling
Spin-orbit coupling during atomic scattering may cause spin-flip
of the photoelectron (Mott scattering). This
relativistic effect can be very important for heavy
atoms and/or high photoelectron energies.
A way to account for this is by reading relativistic scattering phase shifts using the command scatterer i ... Also, for atoms whose scattering properties are defined via their
muffin-tin potentials (either because these potentials have been read
from external data files or because the command muffin-tin has been invoked), the scattering phase shifts are calculated inside
the code, and these phase shifts are relativistic or non-relativistic
depending on the status of the flag scattering-so, which
can be set using the following command:
scattering-so on/off
Scattering phase shifts calculated internally within the code
are relativistic/non-relativistic after this command
is invoked with the on/off option. The relativistic
phase shifts account for spin-flip processes (Mott scattering). Default: scattering-so off.
Effects of atomic magnetic moments
When the atom has a magnetic moment, the exchange and correlation
for electrons with their spin parallel or anti-parallel with respect
to the direction of that moment is different, and therefore, they
are scattered differently (i.e., their scattering must be described
by a different set of phase shifts). The orientation of the atomic
magnetic moments can be specified during the definition of the cluster
using the commands add cluster ....
The following commands can also be used:
orientation θmagnet φmagnet
Reset the orientation of the magnetic moments of
all atoms, so that they will point to the polar
direction (θmagnet, φmagnet). The flag orientation on/off is not affected by this command.
orientation on/off
Enable/disable orientation of target atoms, to be used
for magnetic moments or non-spherical scattering matrices in general.
If on is selected for the first time and
a cluster is already defined, the orientation of
all atoms is set to θmagnet=0 by default
(that is, all atomic magnetic moments point to the positive z direction). Default: orientation off.
Notice that orientation on has to be invoked to activate the orientation of magnetic moments.
| Atomic scattering data-file format |
The external data file that contains the atomic scattering properties of a given atom can contain data of three different kinds:
The scattering phase shifts, which define completely the scattering properties of spherical atoms. These have to be given for an energy range that contains the photoelectron energies that will be eventually used.
The scattering muffin-tin potential for a particular atom. The scattering phase shifts for that atom are calculated only whenever they are needed. The muffin-tin potential is referred to the muffin-tin zero.
The full scattering t-matrix, which can represent non-spherical atoms. Again, this has to be given within a sufficient energy range.
These kinds of data files are considered separately in what follows,
but any of them can be used as input of scatterer i filename.
Atomic scattering phase shifts
The phase-shift data files explained here
represent atoms described by spherically-symmetric
potentials. These can be non-relativistic
phase shifts, relativistic phase shifts, or
even separate sets of phase shifts for spin-up
and spin-down electrons (in that case the
atomic potential including spin interaction is not
really spherical). The general form of these files
is:
| nk units1 lmax regular/ex/so real/complex/tl |
| k1 ps10 ps11 ... |
| k2 ps20 ps21 ... |
| ... |
| knk psnk0 psnk1 ... |
(keywords are given in different color). Here, psj0 ... are phase-shift values (the actual order depends on whether regular, ex, or so is chosen; see below) at the energy (or momentum) kj. The phase shifts are given in radians and any 2π jumps are automatically eliminated while they are read so as to avoid possible interpolation problems (caution: widely separated kj's can be problematic in this respect). nk is the number of energies (or momenta) and their values kj are given in units of units1, either in decreasing or decreasing order. When the keyword real is specified, the phase shifts are assumed to be real, so that psjl is just a real number. However, some absorption during the atomic scattering process can be introduced by using complex phase shifts. This is done by selecting the keyword complex, and then the entry for each phase shift psjl is actually two real numbers: the real part and the imaginary part in that order. If the keyword tl is used instead, each entry is the real part and the imaginary part of the t-matrix element sinδ exp(i δ), where δ is the corresponding phase shift (only the diagonal elements of the scattering matrix are entered). Finally, either regular, or ex, or so must be chosen, meaning:
regular: regular phase shifts; each line contains: δ0 δ1 ... δlmax; if spin-polarized calculations are performed, both electron spin components are scattered using the same set of phase shifts.
ex: the effect of an atomic magnetic moment on the atomic scattering is incorporated by assigning different phase shifts to spin up and spin down electron wave function components; each input line contains: δ0u δ1u ... δlmaxu δ0d δ1d ... δlmaxd, where the superindices u and d refer to spin up and spin down components, respectively.
so: spin-flip processes during photoelectron scattering are incorporated; this is done by using relativistic phase shifts in a two-component spinor representation; then, each line contains: δl=0,j=1/2 δl=1,j=3/2 δl=2,j=5/2 ... 0 δl=1,j=1/2 δl=2,j=3/2 ... Notice that instead of δl=0,j=-1/2, which does not exist, we include a zero.
Here is an example of a phase-shift data file:
| 7 k(A) 4 regular real 3.2382 0.1018 1.0211 1.3299 0.1949 0.0227 3.4346 0.0149 0.9595 1.3810 0.2583 0.0345 3.6204 -0.0613 0.9060 1.4159 0.3244 0.0494 3.7971 -0.1290 0.8594 1.4431 0.3891 0.0676 3.9659 -0.1900 0.8188 1.4672 0.4493 0.0888 4.1279 -0.2457 0.7831 1.4908 0.5028 0.1129 4.2837 -0.2974 0.7513 1.5147 0.5486 0.1393 |
This is another example for nk=1 (then the units
and value of the energy or momentum are not necessary; caution: this
means that these phase shifts are assumed to be appropriate for the
current kinetic energy): 1 3 regular real 0.8 0.6 0.2 0.1.
Atomic muffin-tin potential
The muffin-tin potential can be input to define the
scattering properties of a given atom, which is
considered to be spherically symmetric. Then,
the scattering phase shifts are calculated whenever they
are needed. For an emitter, a screened Coulomb hole
is added to this potential to calculate the
scattering phase shifts of the ionized atom in the solid.
Here is the format of the potential data files:
| potential/potential2 |
| nr Z units2 units1 rV/V |
| r1 a1 [b1] |
| r2 a2 [b2] |
| ... |
| rnr anr [bnr] |
(keywords are given in different color)
where nr is the number of radial points, Z is the atomic number of the atom under consideration, rj is the distance to the atomic nucleus in units
of units2 (given in increasing order),
the potential V(rj)
is expressed in units of units1,
and aj represents
either rjV(rj) (if the label rV is used)
or simply V(rj) (if the label V is used).
If potential2 is used rather than potential2, the values just
explained for the potential stand for spin-up components of the electron
wave function, whereas spin-down components see a different potential
obtained from bj rather than aj.
The potential is assumed to be given with respect to
the muffin-tin zero. A logarithmic radial grid,
denser near the origin, is recommended. The r=0 point must
not be included in the grid.
Atomic scattering t-matrix
The t-matrix contains a full
description of the atomic scattering process.
Two different kinds of scattering t-matrix
files can be read by the program, appropriate
for axially symmetric atoms (axial) and for arbitrarily
shaped atoms (full), respectively. The general format is:
| nk units1 lmax axial/full ns |
| k1 block1 |
| k2 block2 |
| ... |
| knk blocknk |
(keywords are given in different color). As in the case of the scattering phase shifts, units1 and k1 must be omitted when nk=1. The meaning of lmax is the same here. Furthermore, ns specifies the number of components of the electron wave function: 2 if the electron spin is taken into consideration and 1 otherwise. The elements of the t matrix are given in the lmms representation. The information contained within each block (for each given energy/momentum) depends on whether axial or full is chosen. More precisely, each block contains the blue lines generated by the following loops:
axial:
| for s = 1 to ns |
| for l = 0 to lmax |
| for m = -l to l |
| for s'= 1 to ns |
| for l'=|m| to lmax |
| [s] l m [s'] l' m Re{tslm,s'l'm} Im{tslm,s'l'm} |
full:
| for s = 1 to ns |
| for l = 0 to lmax |
| for m = -l to l |
| for s'= 1 to ns |
| for l'= 0 to lmax |
| for m'= -l' to l' |
| [s] l m [s'] l' m' Re{tslm,s'l'm'} Im{tslm,s'l'm'} |
where the notation [s] and [s'] means that s and s' are only written if ns>1.
The components with s different from s' for ns=2 represent spin-flip processes.
In that case, s=1 refers to the spin-up component
with respect to the quantization direction (ms=1/2),
and s=2 refers to the spin-down component
(ms=-1/2).
The quantization direction is the z axis by default,
but it can be changed to an arbitrary direction using the commands orientation ... (see also add cluster ...). Caution: use oriented atoms with axial only (not with full).
| MULTIPLE SCATTERING |
The way multiple scattering is performed has to be specified
by choosing the iteration method and the number of iterations
(see orders ...);
the value of the maximum angular momentum in the multipole
expansion of the wave function around each atomic center of scattering
(see lmax ...);
the value of the muffin-tin zero relative to vacuum
(see V0 ...);
the inelastic mean free path of the photoelectrons;
and the temperature.
The relevant commands are described within this section.
| Multiple scattering parameters |
dmax units2 dmax
Assign the value dmax to the maximum allowed hop distance between cluster atoms
for which the propagation is followed between two successive
scatterings during the MS process. In other words,
the propagation of the electron between one scattering event at
one atom and the next scattering event at another atom is disregarded
when they are separated by a distance larger than dmax.
When the flag cluster surface on has been selected, the distance is redefined in a way similar
to what is described in cluster surface on/off,
so that dmax is compared to the length of the
path followed by
the photoelectron to the atom to which it is propagated
(same as above) plus the distance from the latter to the solid surface. Default: dmax=infinity.
iteration method [η]
Specify the kind of iteration method used to solve
self-consistently the MS equations.
Here, the real parameter η needs to be given only when
the SR method is selected.
The parameter method can be one of the following: Jacobi, SR, recursion, and exact.
(See García de Abajo, Van Hove, and
Fadley, Phys. Rev. B 63, 75404 (2001) for further
details.)
All of these methods take approximately
the same time per iteration,
except the exact method, which takes much longer, since
it performs a direct inversion of the secular MS matrix.
The Jacobi method corresponds to the regular MS series, whereas
the recursion method is much more robust and is guaranteed to
convege (unlike the Jacobi method). Default: Jacobi.
giant-matrix-output filename
Select the exact inversion method (see iteration exact)
and writes the inverted giant matrix to file filename (in consecutive order as it is calculated, only in the next scan ... command). For each energy/momentum,
it writes a first line with the number of atoms N,
the number of wave function components nc, lmax, and the electron (internal) energy (in eV); then,
the cartesian coordinates (x,y,z) of each cluster atom are given
in consecutive order (in a.u.); and finally,
one line with the real and imaginary part of the inverse of the
giant matrix
(the external loop runs over rows, and the internal loop over columns).
The matrix index is i=(lmax+1)2 (a nc + c)
+l2+l+m,
where a, c, l, and m run over atoms, wave function components,
orbital quantum numbers, and azimuthal quantum numbers, respectively.
lmax lmax
Set the maximum (global) effective orbital angular momentum number lmax to be used in scan ... commands,
including scan pd ... for PD calculations. Default: lmax=15.
orders n o1 ... on
Specify orders of iteration to be printed out when invoking scan pd ... (this command is ignored if the exact inversion method is chosen).
The number of orders is n,
and they are given in the list o1 ... on.
Order 0 corresponds to the direct photoelectron wave function,
in the absence of scattering.
Our experience is that
relatively good convergence is typically achieved for
orders in the 10-20 range.
This command has to be invoked with n>0 before
using scan pd ... Default: n=0.
V0 units1 V0
Specify the energy of the vacuum level with respect
to the muffin-tin zero (V0>0) in units given by units1. This determines the
energy of the photoelectrons
inside the solid (i.e., E+V0,
where E is the emission energy relative to vacuum) and
the photoelectron refraction at the surface. Default: V0=0.
| Electron inelastic mean free path |
Defining commands
imfp filename
Read inelastic mean free path from file filename (see format below).
When this command is invoked, all previous information given by
the command Vi ... is lost.
Then, given a certain value of the inelastic mean free path λinel, the program
defines the imaginary part of
the momentum of the photoelectron inside the solid as ki=1/(2 λinel),
where the factor of 2 comes about because ki enters the electron wave function rather than the intensity.
The real part of that momentum is obtained as kr=[2 (E + V0)]1/2 in atomic units,
where E is the photoelectron kinetic energy in the vacuum
and V0 is the so-called muffin-tin zero. Default: infinity.
imfp TPP-2M ρ Nv Ep Eg
Calculate the electron inelastic mean free path using
the approximate TPP-2M formula (see Tanuma, Powell, and Penn, Surf.
Interface Anal. 21, 165 (1994)),
where ρ is the solid density
(in g/cm3), Nv is the number of valence electrons per atom,
Ep is the plasmon frequency in eV, and Eg is the bandgap energy (in eV;
set Eg=0 for metals).
Tabulated values of these parameters for many materials
can be found in Tanuma, Powell, and Penn, Surf. and Interface
Anal. 17, 927 (1991) (see also
Powell and Jablonski, Surf. and Interface Anal. 29, 108 (2000),
and references therein). Default: infinity.
Vi units1 Σi
This is an alternative to the imfp command just described.
It sets the value of the imaginary part of the photoelectron
self energy inside the solid in units of units1.
When this command is given, all previous information introduced
by the commands Vi ... or imfp ... is lost.
Then, the complex momentum of the photoelectron inside
the solid is calculated as k=[2 (E + V0 + i Σi)]1/2 in atomic units,
where E is the photoelectron kinetic energy in the vacuum
and V0 is the muffin-tin zero
relative to vacuum. Default: infinity.
scan imfp filename
Print out electron inelastic mean free path (in Å)
to file filename within the range specified by emission energy ...
clear imfp
Remove stored information on inelastic mean free path.
attenuation-type 0/1
Set to 0 to use hl(+)(kd) inside the free-electron
Green functions or 1 to use hl(+)(krd) exp(-kid) instead,
where k=kr+i ki is the
photoelectron momentum inside the cluster and d is an interatomic distance.
The imaginary part of k is determined by
the electron inelastic mean free path,
or alternatively, by defining an imaginary part
of the inner potential. Default: attenuation-type 0.
Inelastic mean free path data-file format
The general form of the file that contains the
inelastic mean free path (read with the
command imfp filename) is:
| nk units1 units2 |
| k1 imfp1 |
| k2 imfp2 |
| ... |
| knk imfpnk |
where imfp1 ... are the values of the
inelastic mean free path, given in units of units2,
at the nk energies (or momenta) k1 ..., given
in units of units1 in either increasing or decreasing order.
Here is an example:
| 7 E(eV) l(A) 50 7.0 60 6.5 70 6.0 80 5.5 90 6.0 100 6.5 110 7.0 |
This is another example to illustrate the nk=1 case
(the units and values of the energy or momentum are not necessary): 1 l(A) 5 (imfp equal to 5 Å).
| Setting the temperature |
Thermal vibrations create some diffuse scattering, that is, some loss of coherence in the electron wave function. Here, this is accounted for by using temperature-dependent phase shifts (calculated inside the code), which contain a finite imaginary part. A Debye temperature must be also selected, which roughly indicates how rigid is the solid. The temperature is selected using the following commands:
temperature T TDebye
Set the temperature equal to T and the Debye
temperature equal to TDebye, both in Kelvin.
This causes the scattering phase shifts to be changed with
respect to those for T=0, so that the new phase shifts
represent an atom that vibrates isotropically. Pendry's
procedure is used (see Pendry, Low Energy Electron Diffraction,
Academic Press, 1974).
Temperature effects are not implemented for scatterers defined
via their scattering t-matrices. Default: T=0, TDebye=100.
temperature-T T
Set the temperature equal to T in Kelvin. Default: T=0.
Debye-temperature i TDebye
Set the Debye temperature of element i (either an atomic
symbol or an atomic number) to TDebye Kelvin. Each chemical element can have
a different Debye temperature. Notice that the command temperature ... reselts all Debye temperatures. Default: TDebye=100.
The averaging procedure to produce temperature-dependent phase shifts requires to know the atomic mass of the scatterers. The mass is already stored inside the code for cluster atoms that are identified by their atomic numbers of symbols. However, when the scattering properties are defined via external data files using the command scatterer i ..., the mass needs to be introduced by the user via the following command (if no mass is specified, it is assumed to be infinite):
scatterer i mass M
Set the mass of scatterer i,
previously defined using scatterer i ... M can be either a numerical value (the mass itself
in atomic units)
or an atomic symbol. In the latter case, the mass is taken
as that of the most abundant isotope of that particular atomic
species. Default: M=infinite.
| UNITS, COMMENTS, AND MISCELLANEOUS |
In this section, we describe some extra commands and features of
the code.
It is very important in particular to end the execution of the program
after the calculation is performed; this is done by invoking the command end. Also important are the physical units that are used to input and output data.
| Physical units |
All external data are converted into atomic units (a.u.)
inside the program. However, the energy/momentum in the output
of scan ... is given in the
same units as specified in the command emission energy ... Angular magnitudes are in degrees both in input and in output files
(except phase shifts, which are always given in radians).
Two different labels are used throughout this manual to refer
to units used in the input files. Namely:
units1: this refers to energies/momenta input data; also, it determines whether the number to which it applies is an energy or a momentum; it can take one of the following values:
E(eV): energy in eV
E(au): energy in a.u. (Hartrees)
E(Ry): energy in Rydbergs
k(A): momentum in Å-1
k(au): momentum in a.u.
units2: this refers to lengths and can take one of the following values:
l(A): length in Å
l(au): length in a.u.
l(nm): length in nm
Finally, the calculated angle-dependent emission probability is given in
atomic units per steradian and the electric field of the external light is
considered to have a strength of 1 a.u. in magnitude (just for
normalization purposes).
| Comments |
Internal comments
It is possible to incorporate comments which are not printed out in
the output file(s). There are two types of internal comments:
One-line comments, which start with // and finish at the end of the current line.
Those that start with /* and end with */ (they can be multi-line).
External comments
These are comments that are directly printed out in the
output file(s). They start with " and end with " too.
Special characters can be printed using \: \\ to print \, \" to print ", \n to print a line skip.
These comments must be given in between
commands, that is, they cannot be inserted in between
the keywords or parameters of a single command.
| Wave function input/output |
The photoelectron wave function φ(r)
is expressed in a basis set of
spherical harmonics attached to each atom of the cluster:
φ(r) = Σα,lms φα,lms il hl(+)(k|r-Rα|) Ylm(Ωr-Rα) |s,
where s=1 and s=2 stand for spin up and spin down
components.
The wave function is thus completely determined by
the (complex) electron momentum k,
the atomic positions Rα,
and the coefficients φα,lms.
This information can be input and output using the commands explained
below.
Setting the direct wave function for emitter atoms
The code allows one to manually define the direct wave function (i.e., the
excited electron wave function before scattering takes place)
for the emitter atoms, in order to represent arbitrary
emission processes from localized atoms (e.g., PD and Auger emission).
wf manual ns lmax Re{wf1} Im{wf1}
Re{wf2} Im{wf2} ...
Specify the direct wave of the emitters when
the flag wf manual on has been
selected. Non-emitter atoms do not contribute to the direct wave.
For emitter atoms αe,
the coefficients φαe,lms at zeroth order of iteration are assigned
using this command.
These coefficients are represented by the
parameters Re{wf1}, Im{wf1}
Re{wf2}, Im{wf2}, ...
The order in which they are given is the same
as in the blue lines generated by the following loops:
| for s = 1 to ns |
| for l = 0 to lmax |
| for m = -l to l |
| Re{φαe,lms} Im{φαe,lms} |
where ns=2 for spin-polarized calculations and ns=1 otherwise. Notice that lmax can differ from the value selected by the command lmax lmax: the latter refers to MS and can take relatively large values in the 4-20 range, whereas the former is dictated by the selection rules for the excitation process under consideration, (e.g., 2 for photoemission from p states). It is convenient to stress that the emission of different emitter atoms is added up incoherently.
wf manual on/off
Enable/Disable the manual definition of the direct wave function.
When this flag is on, the atoms selected as emitters contribute with a direct wave function (i.e., at zero order of scattering)
specified using the command wf manual ns lmax ... This allows one to consider any kind of electron emission
coming from localized atoms (e.g., Auger emission) by
just introducing by hand the appropriate coefficients. Default: wf manual off.
Reading and writing an entire direct wave function
The entire wave function, along with the cluster,
can also be input and output using the following commands:
wf input ns filename
Read the electron momentum, lmax,
the cluster coordinates,
and the wave function coefficients from file filename.
The number of spin components of the wave function is
given by ns.
This has to be used in combination with the
command scan ms ....
When this latter command is invoked,
all information previously stored on
the cluster,
the list of emitters, and lmax is lost.
The maximum order of iteration introduced by means of the command orders ... must be at least equal to the maximum order
of the stored wave function (see omax below).
The command scan ms ... will evaluate
amplitudes or intensities according to the specifications
given by emission angle ... and orders ....
MS is only calculated for iteration
orders larger than the maximum one of the stored wave
function.
One has to set filename=none in order to
run scan pd ..., in which case scan ms ... cannot be used.
The format of the generated file is explained below; it is compatible with the output
of wf output ... Note: the value of lmax assigned by
a call to the
command lmax lmax before the command scan ms ... is invoked has to be at
least equal to the maximum lmax inside the wave
function data file. Default: filename=none.
wf output filename
Append the (complex) electron momentum k, lmax,
the atomic positions Rα,
and the coefficients φα,lms to file filename.
For filename=none, the wave function is not written to any file.
Otherwise, the electron momentum, the cluster coordinates,
and the wave function are written to the specified file
whenever a MS calculation is performed. This is done for the
complete different combinations of
every electron energy,
every emitter, and
every initial core level and sublevel.
The format of the generated file is explained below; it is compatible to be used as input
of wf input ... Default: filename=none.
The format of the wave function file used as input in this commands
corresponds to the blue lines generated by the following algorithmic
structure (the underlined words are just comments included in the file):
| Momentum (a.u.) Re{k}
Im{k} Maximum order omax lmax_eff lmax Cluster cluster block Coefficients |
| for o = 0 to omax |
| for α = 1 to N |
| order o atom α ns ns lmax l'max |
| for s = 1 to ns |
| for l = 0 to l'max |
| for m = -l to l |
| s l m Re{φαe,lms} Im{φαe,lms} |
Here, l'max refers to the inner loops and can
be different for each atom α;
also l'max cannot be larger than lmax.
The number of atoms N is fixed inside the cluster block; the latter has exactly the
same structure as an atomic cluster data file.
The full wave function data file can contain several wave functions
for different electron momenta or even different atomic clusters.
Each of the wave function data has the structure just specified,
and the entire data file is just a concatenation of all of them.
| Other commands |
Redirecting input and output
include filename
The instructions contained in file filename are
included in the input file currently being interpreted.
The last sentence in that file has to be end,
after which control is returned to the main input file. Synonym: insert.
output filename
Re-direct the output to file filename. After this command is
encountered, the remainder of the output is appended
to file filename until the program
ends or the output is re-directed again.
If filename=terminal, the
output is sent to the standard output device. Default: filename=terminal.
Finishing the execution of the program
end
Terminate the execution of the program. The remainder of
the input file is ignored. Synonyms: exit, quit.
Type of interpolation and tolerance
interpolation linear/spline
Use linear/spline interpolation to interpolate in
momentum/energy when reading phase shifts, etc.
The interpolation mode affects all input data
(phase shifts, etc) both at the moment when they are read and
when they are evaluated (i.e., if some data are read
with the linear interpolation flag then they
will always be evaluated with linear interpolation).
If spline interpolation is selected,
the number of points (i.e., the number of energies or momenta)
must be either 1 (this represents a constant value) or larger than 3.
This limitation does not apply to linear interpolation. Default: spline.
tolerance angles/distances ε
Two angles/distances that differ by less
than ε degrees/a.u. are considered
to be equal inside the code.
This has an effect on the internal evaluation of Green functions
and rotation matrices: if one of those elements is requested
for a given value of a distance or an angle, and another such
element has been calculated and stored already during the
current execution of the program with a value of the distance
or the angle, respectively, that differs less that ε from the requested one, then
the stored element is used. Default: ε=0.003 degrees/0.00005 a.u.
tolerance tmat ε
A scattering matrix element (or phase shift) smaller than ε will be taken to be zero.
This has an immediate effect on the effective value
of lmax, since
the phase shifts become smaller for
increasing values of l: the effective lmax is calculated as that of
the maximum l for which the phase shifts are
non-zero, and the program dynamically evaluates lmax for each atom and each energy.
Notice that the
iteration methods used to solve the multiple scattering
series within the program take a time proportional to
~(lmax+1)3.
Actually, different scattering atoms may have different
effective values of lmax, and this is
used in the evaluation of the multiple scattering series
to reduce the computational demand. Default: ε=0.00005.
Reporting execution time and other comments
time
Print total CPU time and partial CPU time since the last call to time.
verbose on/off
Do/Do not copy output of scan ... commands to standard output. Default: on.
T matrix of the entire atomic cluster
tmat lmax filename
Calculate the t matrix of the entire atomic cluster
and write it to file filename.
Here, lmax is the maximum l up to
which the elements tlm,l'm' are calculated
(this must be generally larger than the value specified by the
command lmax lmax,
which refers to atomic scattering within the cluster; as a first
guess, the value of lmax is given by
the electron momentum times the "radius" of the cluster).
The t matrix is calculated with respect to
a multipole origin given by the reference point
(xr, yr, zr).
The result has the same format as
for atomic
scattering t-matrices, and in particular it
will be labeled full (see format).
Actually, it can be used as input of scatterer i filename.
This command takes a very long time when more than a few atoms
are considered. For a similar command to output phase shifts of
individual atoms, see scan scatterer i phase-shifts ...
| Miscellaneous |
Inline specifications
Whenever an input file is expected (e.g., after cluster input filename, scatterer i filename, etc.)
one can type inline and then the rest of
the information required by the current command is read
from the main input file. Example: imfp inline 1 l(A) 6,
which specifies an imfp of 6 Å.
Surface position and z axis
The polar angles are generally given with respect to the positive z-axis, which points perpendicularly to the surface in
the outward direction. The position of the latter is fixed by the command cluster surface zsurf, so that the
electronic surface is considered to be at z=zsurf.
Miscellaneous keywords
The words infinite and zero can be used in the
input files meaning an arbitrarily large number and 0, respectively.
Output file names
Some of the commands require an output file name parameter
(filename). The output of those commands is written into their
respective output file names, except when filename=none, in which case no output file is created.
For some commands, filename=terminal redirects the output
to the standard output device (the terminal).
The output is sent to the standard output device
(the screen) in any case, unless the command verbose off has been invoked.
| ELECTRON DIFFRACTION CALCULATION |
Once all necessary input has been provided,
electron diffraction (e.g., PD) calculations can be performed
by invoking the commands explained next
(e.g., scan pd ... for PD).
Information on some internal statistics of the calculation
can then be obtained by using the commands report ... discussed below.
The code has the capability to perform both spin-independent and
spin-polarized MS calculations. The latter takes approximately
twice the time as compared with the former (under otherwise identical
conditions), since two spin components have to be propagated, rather
than one (the bulk of the computation time is invested in
propagating the wave function by means of the free-electron
Green function between consecutive atomic scattering events).
Spin-polarized calculations are performed whenever one of
the following conditions is fulfilled:
Some scatterer is defined via relativistic scattering phase shifts read from an external file using the command scatterer i ....
Some scatterer is defined via its muffin-tin potential and the command scattering-so on has been invoked. The muffin-tin potential comes about because either it has been read from an external file using scatterer i ... or because the command muffin-tin has been used.
Some scatterer is defined via its scattering phase shifts read from an external file using the command scatterer i ... and incorporating magnetic exchange effects.
Some scatterer is defined via its t-matrix, read from an external file using the command scatterer i ..., and it has two spin components (ns=2).
Separate radial matrix elements for the two spin components have been specified for the emitter(s) and they have been introduced via the command rmat filename. (Only for scan pd ...)
Non-relativistic, spin-independent radial matrix elements have been specified for the emitter(s) (either by reading them via the command rmat filename or by calculating them inside the code for emitters defined by their muffin-tin potentials), but magnetic sublevels of emission have been selected using initial j0/mj0. (Only for scan pd ...)
The unscattered wave function defined for the emitter by means of the command wf manual ... has two spin components (ns=2). (Only for scan pd ...)
The (unscattered) wave function is read from an external file using the command wf input ... and it has two independent spin components (ns=2). (Only for scan ms ...)
In that case, one can select the direction on which the detector
projects the electron spin (for spin-polarized detectors) using
the commands spin ...
If none of the above conditions if fulfilled, the program performs
spin-independent MS calculations and the command spin ... is ignored.
Finally, photoelectron intensities can be averaged over a final
acceptance-angle window using the command emission angle window ... This can be used to represent a finite angular width in the
photoelectron detector.
Notice however that this command is ignored when
any of the options integrated, half-integrated,
or amplitude is used in the commands explained below.
Photoelectron diffraction
scan [integrated/half-integrated] pd
[amplitude] filename
Scan PD intensities according to the
parameters previously specified and write the results
in file filename.
Both surface refraction and transmission effects are incorporated
through a barrier potential V0.
If the option amplitude is included,
complex photoelectron wavefunction
amplitudes (at the detector position)
rather than intensities are printed out.
If the option integrated is included,
the resulting intensity is angle-integrated within the range defined by emission angle ... With half-integrated, the result is integrated
in the azimuthal direction only, so that θ-dependent results are given.
The keywords amplitude and integration cannot
be given simultaneously, since it does not make sense to
integrate amplitudes. Also, the keyword amplitude can only be used with only one emitter atom in the cluster
and one individual core sublevel (e.g., the one specified
by l0 1 m0 0 ms0 1 in the lmms representation).
Diffraction of predefined wave
scan [integrated/half-integrated] ms nwf [amplitude] filename
The same as scan pd ...,
except that the wave function is read from an input file
as specified by the command wf input ....
The parameter nwf indicates the
number of different wave function data sets
that must be used from the file filename read by wf input filename.
The wave function does not have to represent a photoelectron
necessarily.
This command can be used to calculate MS effects on
arbitrary wave functions generated by a number of
different physical processes. Note: this command destroys any information previously
stored on the cluster or the list of emitters; also, it
deactivates the flag wf input ..., so that the
wave function data file has to be specified again in subsequent
calls to scan ms ...
Reporting information on diffraction calculations
report links
Print number of bond distances and bond
angles that have been used in the last call to scan pd ...
report lmax
Print effective value of lmax used in the last run of scan pd ....
report size
Print information on lmax and the number of
non-zero elements in Green functions
and rotations used in the last call to scan pd ...
| LIST OF COMMANDS IN ALPHABETICAL ORDER |
Note. Keywords in square brackets are optional and keywords separated by / are multi-choice (select only one of them). The parameter sector of the commands (i.e., numbers, units, filenames, etc.) is written in blue.